
Sickle cell disease, also termed sickle cell anemia, is a genetic disorder in which hypoxia, or oxygen deprivation, causes red blood cells (erythrocytes) to assume a rigid sickle shape, blocking blood flow within blood vessels. The disease results from a single nucleotide mutation in the gene for beta-globin (β-globin), which causes production of an abnormal form of hemoglobin—an oxygen-carrying protein in red blood cells. Millions of people worldwide suffer from sickle cell disease, enduring lifelong anemia, as well as possible organ damage and potentially early death. The abnormal rigidity of sickle cells also causes painful obstruction of small blood vessels, and recurrent painful episodes are frequent in the absence of proper therapies. The most common treatment options include analgesics for pain, antibiotics for fighting infections, transfusions for episodes of severe anemia, and hydroxyurea (an oral medication that hinders formation of sickle-shaped red blood cells and reduces inflammation). See also: Anemia; Blood; Blood vessel; Hemoglobin; Human genetics; Mutation; Nucleotide; Sickle cell disease
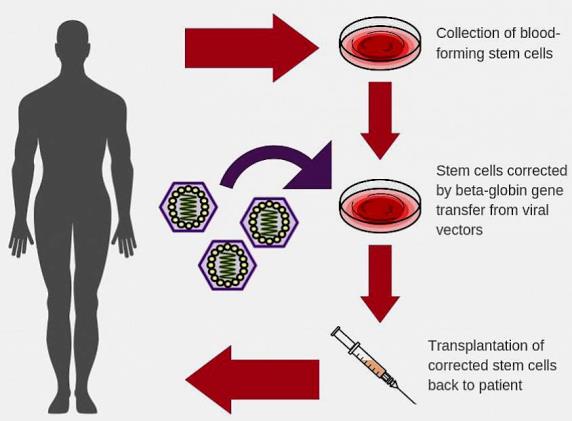
Recently, advances in stem cell transplantation methods and gene therapies have led to novel approaches to treat and possibly cure sickle cell disease. For example, in one current treatment option, blood-producing (hematopoietic) bone marrow cells of a patient with sickle cell disease are destroyed and replaced with stem cells drawn from the bone marrow of a healthy relative unaffected by the sickling trait. However, in many cases, a healthy donor is not available for this procedure. Therefore, researchers are utilizing various gene therapy techniques to alter a patient's own bone marrow stem cells. In this technique, scientists add a normal copy of the beta-globin gene to stem cells through the use of a viral vector—that is, a virus-based agent that delivers therapeutic genetic material into cells. Then, a physician transplants the corrected stem cells into the patient's bone marrow, prompting the marrow to produce normal-shaped, functional red blood cells. In animal models, the viral vectors have been highly efficient at incorporating corrective genes into bone marrow stem cells. These vectors remained in place for four years after transplantation. Results are promising, and clinical trials of the viral vectors in humans are expected to begin. See also: Bone; Hematopoiesis; Stem cells; Transplantation biology
Another notable treatment idea involves direct gene editing, which would enable in vivo (taking place inside a living cell or organism) correction of the gene mutation responsible for sickle cell disease. The use of direct gene editing, particularly utilizing CRISPR technology, would avoid the difficulties involved in transplantation of ex vivo modified stem cells. (CRISPR is a genome-engineering technique that allows precise targeting of specific stretches of genetic code and editing of DNA at designated locations.) However, safety concerns related to both genotoxic effects and low levels of efficiency must be addressed before clinical testing of this concept can begin. In addition, the cost of direct gene editing is substantial, and the ability to utilize such an expensive therapeutic approach in many areas of the world that can least afford this option is an impediment to its actual use. Therefore, investigators are still focused on finding inexpensive drugs that could prevent the sickling of red blood cells or allow red blood cells to transit the vessel microcirculation before sickling occurs. See also: CRISPR/Cas9 gene editing; Deoxyribonucleic acid (DNA); Gene; Genetic code; Genetic engineering

